Benzylic activation and stereocontrol in tricarbonyl(arene)chromium complexes
Benzylic activation and stereocontrol in tricarbonyl(arene)chromium complexes refers to the enhanced rates and stereoselectivities of reactions at the benzylic position of aromatic rings complexed to chromium(0) relative to uncomplexed arenes. Complexation of an aromatic ring to chromium stabilizes both anions and cations at the benzylic position and provides a steric blocking element for diastereoselective functionalization of the benzylic position. A large number of stereoselective methods for benzylic and homobenzylic functionalization have been developed based on this property.[1]
Introduction
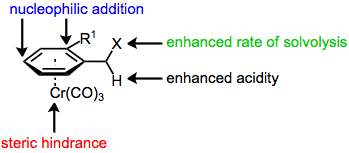
The reaction between tricarbonylchromium complexes Cr(CO)3L3 and electron-rich or electron-neutral aromatic rings produces tricarbonyl(arene)chromium complexes (arene)Cr(CO)3.[2] Complexation to chromium(0) activates the side chain of the arene, facilitating dissociation of a benzylic proton or leaving group or nucleophilic addition to the homobenzylic position of styrenes. Further transformations of the resulting conformationally restricted, benzylic anion or cation involve the approach of reagents exo to the chromium fragment. Thus, benzylic functionalization reactions of planar chiral chromium arene complexes are highly diastereoselective. Additionally, the chromium tri(carbonyl) fragment can be used as a blocking element in addition reactions to ortho-substituted aromatic aldehydes and alkenes. An ortho substituent is necessary in these reactions to restrict conformations available to the aldehyde or alkene.[3] Removal of the chromium fragment to afford the metal-free functionalized aromatic compound is possible photolytically[4] or with an oxidant.[5]
(1)

Mechanism and stereochemistry
Prevailing mechanism
The majority of benzylic functionalization reactions of tricarbonyl(arene)chromium complexes proceed by mechanisms analogous to those followed by the free arenes. There are three important differences, however. First, the aromatic ring and benzylic position are activated towards solvolysis, deprotonation, and nucleophilic attack (at the ortho and para positions of the arene) upon complexation to chromium, which is able to stabilize developing charges in the arene ligand. As a result, these reactions of chromium arene complexes are often faster than analogous reactions of free arenes.[6]
(2)
Second, in benzylic cations and anions of chromium arene complexes, rotation about the bond connecting the benzylic carbon and aromatic ring is severely restricted. This bond possesses a significant amount of double bond character due to the delocalization of charge into the aromatic ring (and the stabilization of that charge by chromium).[7]
(3)
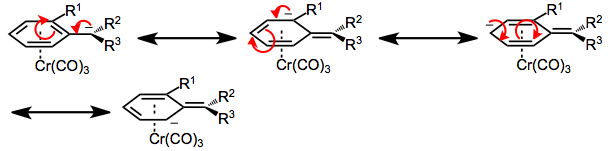
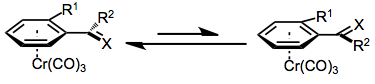
Finally, the chromium tri(carbonyl) moiety serves as a sterically bulky group in reactions of arene chromium complexes, preventing the approach of a reagent endo to chromium. In addition, ortho-substituted aromatic aldehydes and styrenes prefer to adopt a conformation in which the doubly bound oxygen or carbon is pointed away from the ortho substituent. As a result, only one face of the double bond is exposed on the exo face of the aromatic ring. If this were not the case, addition to styrenes and aromatic aldehydes would not be diastereoselective, despite the presence of the chromium tri(carbonyl) group. The ortho substituent is necessary for high stereoselectivity; meta-substituted arenes exhibit very low diastereoselectivity.[3]
(4)
Enantioselective variants
Enantioselective benzylic functionalization methods use the complexed chromium tri(carbonyl) moiety essentially as a chiral auxiliary.[8] Approach of the functionalizing reagent anti to the chromium tri(carbonyl) fragment leads to a single diastereomer of the product complex. After removal of the chromium group with light[4] or an oxidizing agent such as iodine,[5] nearly enantiopure product remains. See the Scope and Limitations section below for several methods for diastereoselective benzylic functionalization.
Scope and limitations
Enantioselective benzylic functionalization reactions depend on the use of enantiomerically pure, planar chiral chromium arene complexes. This section describes methods for the enantioselective synthesis of planar chiral chromium arene complexes, then outlines methods for functionalization of both sp2- and sp3-hybridized benzylic positions.
Preparation of enantiopure, planar chiral chromium complexes
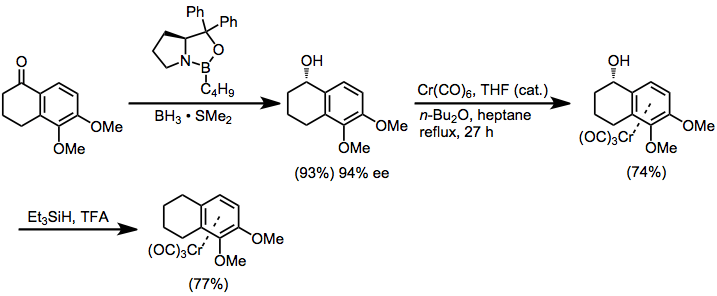
Enantiopure, planar chiral chromium arene complexes can be synthesized using several strategies. Diastereoselective complexation of a chiral, non-racemic arene to chromium is one such strategy. In the example in equation (5), enantioselective Corey-Itsuno reduction[9] sets up a diastereoselective ligand substitution reaction. After complexation, the alcohol is reduced with triethylsilane.[10]
(5)
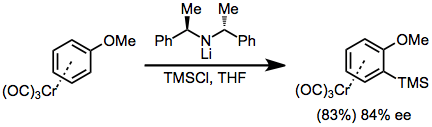
A second strategy involves enantioselective ortho-lithiation and in situ quenching with an electrophile. Isolation of the lithium arene and subsequent treatment with TMSCl led to lower enantioselectivities.[11]
(6)
Site-selective conjugate addition to chiral aryl hydrazone complexes can also be used for the enantioselective formation of planar chiral chromium arenes. Hydride abstraction neutralizes the addition product, and treatment with acid cleaves the hydrazone.[12]
(7)
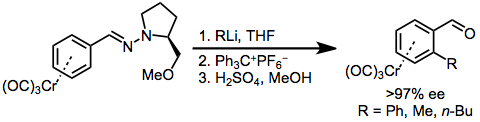
Benzylic functionalization reactions
ortho-Substituted aryl aldehyde complexes undergo diastereoselective nucleophilic addition with organometallic reagents[13] and other nucleophiles. Equation (8) is an example of a diastereoselective Morita-Baylis-Hillman reaction.[14]
(8)

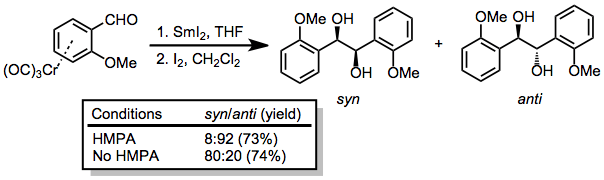
Pinacol coupling and the corresponding diamine coupling[15] are possible in the presence of a one-electron reducing agent such as samarium(II) iodide.[16]
(9)
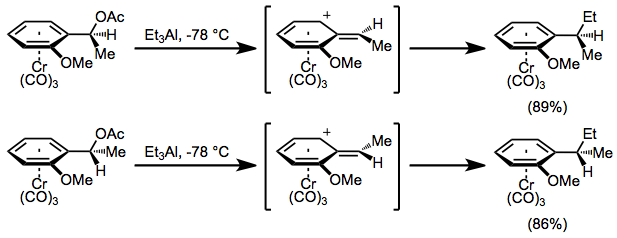
Benzylic cations of chromium arene complexes are conformationally stable, and undergo only exo attack to afford SN1 products stereospecifically, with retention of configuration.[7] Propargyl[17] and oxonium[18] cations undergo retentive substitution reactions, and even β carbocations react with a significant degree of retention.[19]
(10)
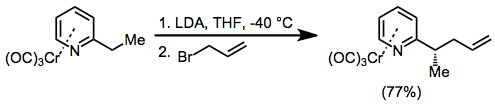
Benzylic anions of chromium arene complexes exhibit similar reactivity to cations. They are also conformationally restricted and undergo substitution reactions with retention of stereochemistry at the benzylic carbon. In the example below, complexation of the pyridine nitrogen to lithium is essential for high stereoselectivity.[20]
(11)
Nucleophilic addition to styrenes followed by quenching with an electrophile leads to cis products with essentially complete stereoselectivity.[21]
(12)

Diastereoselective reduction of styrenes is possible with samarium(II) iodide. A distant alkene is untouched during this reaction, which provides the reduced alkylarene product in high yield.[22]
(13)

Complexation of a haloarene to chromium increases its propensity to undergo oxidative addition.[23] Suzuki cross coupling of a planar chiral chromium haloarene complex with an aryl boronic acid is thus a viable method for the synthesis of axially chiral biaryls. In the example below, the syn isomer is formed in preference to the anti isomer; when R2 is the formyl group, the selectivity reverses.[24]
(14)

Tetralones complexed to chromium may be deprotonated without side reactions. Alkylation of the resulting enolate proceeds with complete diastereoselectivity to afford the exo product.[25]
(15)

Synthetic applications
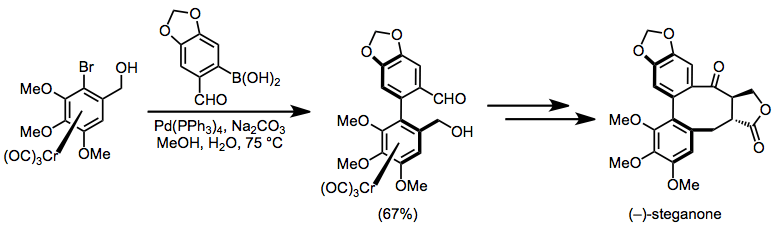
A key step in the synthesis of (–)-steganone involves the enantioselective cross-coupling of a chromium haloarene complex with a boronic acid to afford an axially chiral intermediate with a configuration that matched the target. Elaboration of the arene side chains then afforded that of the target.[26]
(16)
Experimental conditions and procedure
Typical conditions
Tricarbonyl(arene) chromium complexes are susceptible to oxidation in solution and should be handled in an inert atmosphere. Compounds of chromium are toxic and must be used in a well-ventilated fume hood. The aqueous layers from the chromium-mediated reactions and any other waste materials should be disposed of properly. A very simple procedure for decomplexation involves exposure of a solution of the chromium arene complex to sunlight until it becomes colorless. Filtration and evaporation of the solvent provides the chromium-free aromatic compound.[1]
Example procedure[27]
(17)
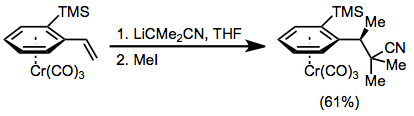
To a cooled (–78°) solution of n-BuLi (1.5 M in hexane, 1.75 mL, 2.63 mmol) in THF (8 mL) was added dropwise with stirring isobutyronitrile (182 mg, 2.63 mmol). The reaction mixture temperature was increased to –20° over 30 minutes. To this solution was added (–)-(1S,2R)-tricarbonyl(2-trimethylsilylstyrene)chromium (674 mg, 1.88 mmol) in THF (5 mL) at –78°. The reaction mixture was brought to –20° quickly and subsequently stirred for 2 hours as it warmed from –20 to 0°. After cooling again to –78°, methyl iodide (441 mg, 3.10 mmol) was added dropwise. After warming from –20 to 0° with stirring over 2 hours, the reaction mixture was left to reach room temperature. The solvent was removed under reduced pressure and the residue dissolved in Et2O (30 mL), washed with saturated aqueous NH4Cl solution (10 mL) and brine (10 mL), dried over MgSO4, and concentrated. Purification by column chromatography (SiO2, light petroleum/ CH2Cl2 2:1 to 1:1) followed by crystallization (hexane/ CH2Cl2), yielded yellow crystals of the title compound (453 mg, 61%), mp 112–114°; [α]23D – 25.6° (c 1.36, CHCl3); IR (CH2Cl2) 1963, 1885 cm−1; 1H NMR δ 0.41 (s, 9H), 1.45 (d, J = 6 Hz, 3H), 1.46 (s, 3H), 1.49 (s, 3H), 1.88 (dd, J = 10, 14 Hz, 1H), 1.95 (dd, J = 3, 14 Hz, 1H), 2.89 (dqd, J = 3, 6, 10 Hz, 1H), 4.98 (dd, J = 1, 6 Hz, 1H), 5.19 (dt, J = 1, 6 Hz, 1H), 5.44 (dd, J = 1, 6 Hz, 1H), 5.57 (dt, J = 1, 6 Hz, 1H); 13C NMR δ 25.5, 25.8, 30.0, 30.6, 34.0, 45.7, 88.4, 90.4, 95.4, 99.0, 100.4, 125.4, 127.4, 233.2.
References
- 1 2 Uemura, M. Org. React. 2006, 67, 217. doi:10.1002/0471264180.or067.02
- ↑ Mahaffy, C. A. L.; Pauson, P. Inorg. Synth. 1990, 28, 137.
- 1 2 Besançon, J.; Tirouflet, J.; Card, A.; Dusausoy, Y. J. Organomet. Chem. 1973, 59, 267.
- 1 2 Mishchenko, O. G.; Klementeva, S. V.; Maslennikov, S. V.; Artemov, A. N.; Spirina, I. V. Rus. J. Gen. Chem. 2006, 76, 1907.
- 1 2 Semmelhack, M. F.; Zask, A. J. Am. Chem. Soc. 1983, 105, 2034.
- ↑ Holmes, J. D.; Jones, D. A. K.; Pettit, R. J. Organomet. Chem. 1965, 4, 324.
- 1 2 Davies, S. G.; Donohoe, T. J. Synlett 1993, 323.
- ↑ Uemura, M.; Hayashi, Y.; Hayashi, Y. Tetrahedron: Asymmetry, 1993, 4, 2291.
- ↑ Itsuno, S. Org. React. 1998, 52, 395.
- ↑ Schmalz, H.-G.; Arnold, M.; Hollander, J.; Bats, J. W. Angew. Chem. Int. Ed. Engl. 1994, 33, 109.
- ↑ Price, D. A.; Simpkins, N. S.; MacLeod, A. M.; Watt, A. P. J. Org. Chem. 1994, 59, 1961.
- ↑ Kündig, E. P.; Liu, R.; Ripa, A. Helv. Chim. Acta 1992, 75, 2657.
- ↑ Bitterwolf, T. E.; Dai, X. J. Organomet. Chem. 1992, 440, 103.
- ↑ Kündig, E. P.; Xu, L. H.; Romanens, P.; Bernardinelli, G. Tetrahedron Lett. 1993, 34, 7049.
- ↑ Davies, S. G.; Donohoe, T. J.; Williams, J. M. J. Pure Appl. Chem. 1992, 64, 379.
- ↑ Taniguchi, N.; Kaneta, N.; Uemura, M. J. Org. Chem. 1996, 61, 6088.
- ↑ Müller, T. J. J.; Netz, A. Tetrahedron Lett. 1999, 40, 3145.
- ↑ Davies, S. G.; Newton, R. F.; Williams, J. M. J. Tetrahedron Lett. 1989, 30, 2967.
- ↑ Merlic, C. A.; Miller, M. M. Organometallics 2001, 20, 373.
- ↑ Davies, S. G.; Shipton, M. R. J. Chem. Soc., Chem. Commun. 1990, 1780.
- ↑ Majdalani, A.; Schmalz, H.-G. Tetrahedron Lett. 1997, 38, 4545.
- ↑ Schmalz, H.-G.; Siegel, S.; Bernicke, D. Tetrahedron Lett. 1998, 39, 6683.
- ↑ Widdowson, D. A.; Wilhelm, R. Chem. Commun. 1999, 2211.
- ↑ Kamikawa, K.; Watanabe, T.; Uemura, M. J. Org. Chem. 1996, 61, 1375.
- ↑ Davies, S. G.; Coote, S. J.; Goodfellow, C. L. In Advances in Metal-Organic Chemistry; Liebeskind, L.S., Ed.; JAI Press: Greenwich, 1991; Vol. 2. pp. 1–48.
- ↑ Monovich, L. G.; Le Huérou, Y.; Ronn, M.; Molander, G,. A. J. Am. Chem. Soc. 2000, 122, 52.
- ↑ Gibson, S. E.; Gil, R.; Prechtl, F.; White, A. J. P.; Williams, D. J. J. Chem. Soc., Perkin Trans. 1 1996, 1007.