Biosynthesis of cocaine
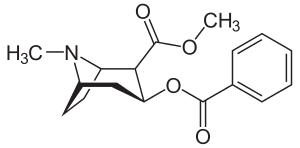
The first synthesis and elucidation of the cocaine molecule was by Richard Willstätter in 1898.[1] Willstätter's synthesis derived cocaine from tropinone. Since then, Robert Robinson and Edward Leete have made significant contributions to the mechanism of the synthesis.
Biosynthesis of N-methyl-pyrrolinium cation
The biosynthesis begins with L-Glutamine, which is derived from L-ornithine in plants. The major contribution of L-ornithine and L-arginine as a precursor to the tropane ring was confirmed by Edward Leete.[2] Ornithine then undergoes a PLP-dependent decarboxylation to form putrescine. In animals, however, the urea cycle derives putrescine from ornithine. L-ornithine is converted to L-arginine,[3] which is then decarboxylated via PLP to form agmatine. Hydrolysis of the imine derives N-carbamoylputrescine followed with hydrolysis of the urea to form putrescine. The separate pathways of converting ornithine to putrescine in plants and animals have converged. A SAM-dependent N-methylation of putrescine gives the N-methylputrescine product, which then undergoes oxidative deamination by the action of diamine oxidase to yield the aminoaldehyde. Schiff base formation confirms the biosynthesis of the N-methyl-Δ1-pyrrolinium cation.
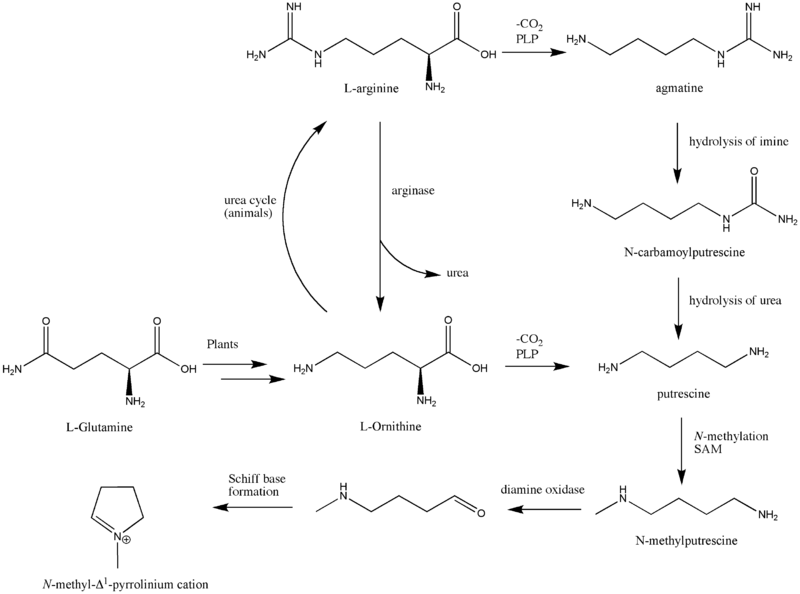
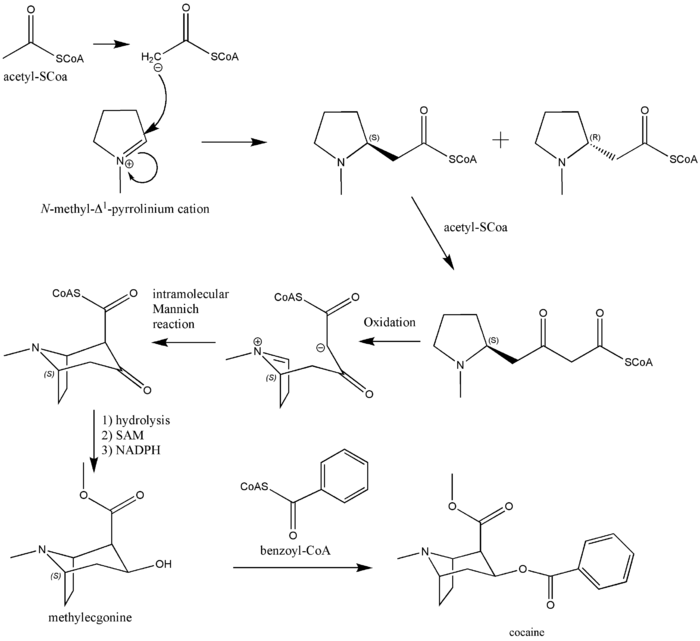
Biosynthesis of cocaine
The additional carbon atoms required for the synthesis of cocaine are derived from acetyl-CoA, by addition of two acetyl-CoA units to the N-methyl-Δ1-pyrrolinium cation.[4] The first addition is a Mannich-like reaction with the enolate anion from acetyl-CoA acting as a nucleophile towards the pyrrolinium cation. The second addition occurs through a Claisen condensation. This produces a racemic mixture of the 2-substituted pyrrolidine, with the retention of the thioester from the Claisen condensation. In formation of tropinone from racemic ethyl [2,3-13C2]4(Nmethyl- 2-pyrrolidinyl)-3-oxobutanoate there is no preference for either stereoisomer.[5] In the biosynthesis of cocaine, however, only the (S)-enantiomer can cyclize to form the tropane ring system of cocaine. The stereoselectivity of this reaction was further investigated through study of prochiral methylene hydrogen discrimination.[6] This is due to the extra chiral center at C-2.[7] This process occurs through an oxidation, which regenerates the pyrrolinium cation and formation of an enolate anion, and an intramolecular Mannich reaction. The tropane ring system undergoes hydrolysis, SAM-dependent methylation, and reduction via NADPH for the formation of methylecgonine. The benzoyl moiety required for the formation of the cocaine diester is synthesized from phenylalanine via cinnamic acid.[8] Benzoyl-CoA then combines the two units to form cocaine.
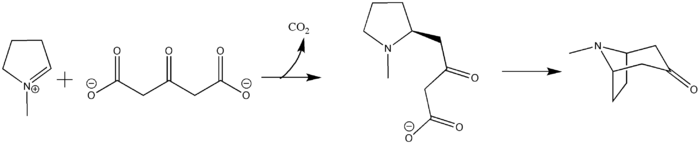
Robert Robinson's acetonedicarboxylate
The biosynthesis of the tropane alkaloid, however, is still uncertain. Hemscheidt proposes that Robinson's acetonedicarboxylate emerges as a potential intermediate for this reaction.[9] Condensation of N-methylpyrrolinium and acetonedicarboxylate would generate the oxobutyrate. Decarboxylation leads to tropane alkaloid formation.
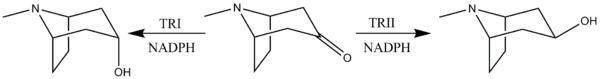
Reduction of tropinone
The reduction of tropinone is mediated by NADPH-dependent reductase enzymes, which have been characterized in multiple plant species.[10] These plant species all contain two types of the reductase enzymes, tropinone reductase I and tropinone reductase II. TRI produces tropine and TRII produces pseudotropine. Due to differing kinetic and pH/activity characteristics of the enzymes and by the 25-fold higher activity of TRI over TRII, the majority of the tropinone reduction is from TRI to form tropine.[11]
References
- ↑ Humphrey AJ, O'Hagan D (October 2001). "Tropane alkaloid biosynthesis. A century old problem unresolved". Nat Prod Rep. 18 (5): 494–502. doi:10.1039/b001713m. PMID 11699882.
- ↑ Leete E, Marion L, Sspenser ID (October 1954). "Biogenesis of hyoscyamine". Nature. 174 (4431): 650–1. Bibcode:1954Natur.174..650L. doi:10.1038/174650a0. PMID 13203600.
- ↑ Robins RJ, Waltons NJ, Hamill JD, Parr AJ, Rhodes MJ (October 1991). "Strategies for the genetic manipulation of alkaloid-producing pathways in plants". Planta Med. 57 (7 Suppl): S27–35. doi:10.1055/s-2006-960226. PMID 17226220.
- ↑ Dewick, P. M. (2009). Medicinal Natural Products. Chicester: Wiley-Blackwell. ISBN 978-0-470-74276-1.
- ↑ R. J. Robins; T. W. Abraham; A. J. Parr; J. Eagles; N. J. Walton (1997). "The Biosynthesis of Tropane Alkaloids in Datura stramonium: The Identity of the Intermediates between N-Methylpyrrolinium Salt and Tropinone". J. Am. Chem. Soc. 119 (45): 10929. doi:10.1021/ja964461p.
- ↑ Hoye TR, Bjorklund JA, Koltun DO, Renner MK (January 2000). "N-methylputrescine oxidation during cocaine biosynthesis: study of prochiral methylene hydrogen discrimination using the remote isotope method". Org. Lett. 2 (1): 3–5. doi:10.1021/ol990940s. PMID 10814231.
- ↑ E. Leete; J. A. Bjorklund; M. M. Couladis & S. H. Kim (1991). "Late intermediates in the biosynthesis of cocaine: 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoate and methyl ecgonine". J. Am. Chem. Soc. 113 (24): 9286. doi:10.1021/ja00024a039.
- ↑ E. Leete; J. A. Bjorklund & S. H. Kim (1988). "The biosynthesis of the benzoyl moiety of cocaine". Phytochemistry. 27 (8): 2553. doi:10.1016/0031-9422(88)87026-2.
- ↑ T. Hemscheidt; Vederas, John C. (2000). Leeper, Finian J.; Vederas, John C., eds. "Tropane and Related Alkaloids". Top. Curr. Chem. Topics in Current Chemistry. 209: 175. doi:10.1007/3-540-48146-X. ISBN 978-3-540-66573-1.
- ↑ A. Portsteffen; B. Draeger & A. Nahrstedt (1992). "Two tropinone reducing enzymes from Datura stramonium transformed root cultures". Phytochemistry. 31 (4): 1135. doi:10.1016/0031-9422(92)80247-C.
- ↑ Boswell HD, Dräger B, McLauchlan WR, et al. (November 1999). "Specificities of the enzymes of N-alkyltropane biosynthesis in Brugmansia and Datura". Phytochemistry. 52 (5): 871–8. doi:10.1016/S0031-9422(99)00293-9. PMID 10626376.