Cerebroretinal microangiopathy with calcifications and cysts
| Cerebroretinal microangiopathy with calcifications and cysts | |
|---|---|
|
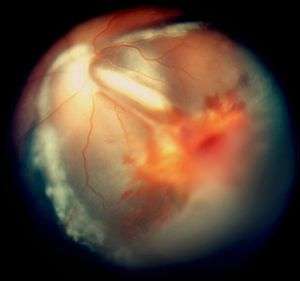
Vitreous hemorrhage and exudative retinal detachment resembling Coats' disease in a child with cerebroretinal microangiopathy with calcifications and cysts | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q87.8 |
| ICD-9-CM | 759.89 |
| OMIM | 612199 |
Cerebroretinal microangiopathy with calcifications and cysts (CRMCC) is a rare genetic disorder, which affects multiple organs.[1][2] Its hallmarks are widespread progressive calcifications, cysts and abnormalities of the white matter of the brain, usually occurring together with abnormalities of the blood vessels of the retina. Additional features include poor prenatal growth, preterm birth, anemia, osteopenia and bone fractures,[3][4][5] and gastrointestinal bleeding.[6] It is caused by compound heterozygous mutations in the conserved telomere maintenance component 1 (CTC1) gene,[7][8] but its exact pathophysiology is still not well understood.
Cerebroretinal microangiopathy with calcifications and cysts is alternatively known as the Coats plus syndrome,[3] a reference to its most typical ocular phenotype.
Presentation
Before birth
A child who has inherited this disorder will grow slower than normal in the uterus before birth and typically will be born before term.[1][2][3] The pregnancy can be complicated by gestational hypertension and pre-eclampsia.
After birth
The majority of affected children present with symptoms and signs relating to the eyes such as leukokoria, redness, irritation and impaired vision, which result from retinal detachment and glaucoma.[1][2][3] A minority present with seizures or spasticity. The time of onset of symptoms varies from infancy to adolescence. Because a child born before term will need to undergo screening for retinopathy of prematurity, some will have abnormalities in their retinal blood vessels detected when they have no symptoms yet.
Signs and Symptoms
Brain
Neurologic symptoms and signs vary depending on the site of the brain abnormalities. Common symptoms are partial epilepsy, asymmetric spasticity, ataxia and cognitive impairment.[1][2][3] The latter affects visuospatial and visuoconstructive skills first. The intracranial pressure can be elevated if cysts develop in the brain. Migraine-like headaches can occur.
Eyes
Smaller blood vessels of the retina are abnormally developed and appear tortuous and dilated to a variable extent, typically in one sector and mainly in the peripheral and temporal portions of the retina.[1][2][3][9] This is known as telangiectasia. The vessel walls are week and leak blood plasma and lipid within and underneath the retina. This leakage can lead to exudative retinal detachment, also known as exudative retinopathy in this context. The detachment typically has a yellowish tint because the fluid under the retina contains lipid. These findings mimic Coats disease.[9] Characteristically, the abnormal vessels are localized and the retinal blood vessels peripheral to the abnormal ones seemingly have failed to develop and are thus not seen.
In some eyes, retinal vessels form small nodules on the surface of the retina, known as angiomas.[1] These can bleed and be attached to the vitreous humour. The attachment can cause traction retinal detachment.
Gastrointestinal tract
Recurrent intestinal bleeding is fairly common.[1][2][6] It originates from telangiectatic small blood vessels in the intestinal mucosa. Additional findings in some individuals are portal hypertension and liver failure.[1][2]
Blood
Many affected children develop anemia, which may be macrocytic in type.[1][2][4] Some also develop thrombocytopenia. Bone marrow examinations may show megaloblasts and increased erythropoiesis or bone marrow suppression.[1][2][4]
Bones
The long bones show osteopenia and pathologic fractures can occur.[1][2][4][5]
Skin, nails and hair
Some children have sparse and greying hair, café au lait spots and nail dystrophy.[1][2][9]
General
Most patients continue to grow poorly after birth.[1][3]
Genetics
Typical childhood-onset cerebroretinal microangiopathy with calcifications and cysts is caused by compound heterozygous mutations in the conserved telomere maintenance component 1 (CTC1) gene[10] located in chromosome 17p.31.[7][8] A late-onset phenotype without abnormal eye findings from a CTC1 mutation has been reported.[8]
CTC1[10] is a component of the CST complex,[11] which is additionally composed of oligonucleotide/oligosaccharide-binding fold containing 1 (coded by OBFC1,[12] also known as STN1) and telomerase capping complex subunit homolog 1 (coded by TEN1).[13] CST complex is evolutionarily conserved.[11] It binds to single-stranded DNA and associates with a fraction of telomeres, potentially protecting them.
Pathophysiology
Angiomas and numerous abnormal, small, dilated telangiectatic vessels with thickened, sclerotic and calcified walls have been found in those brain areas which also show calcifications.[1][2]
By analogy to Coats disease, the exudative retinopathy is thought to result from breakdown of the blood-retinal barrier at the level of the vascular endothelial cell, resulting in leakage of blood plasma and lipid.[14] Macrophages then migrate into the retina and subretinal space and digest the lipid. The accumulation of the proteinaceous exudate and macrophages thickens the retina, leading to exudative retinal detachment.
Diagnosis
Clinical
The retinal changes are easily identified by ophthalmoscopy, which is performed under general anesthesia if the child is very young. The abnormal vessels are even better seen with fluorescein angiography. In advanced disease, glaucoma is diagnosed by measuring intraocular pressure and cataract by using slit lamp biomicroscopy.
Imaging findings
The most consistent finding are widespread calcifications, which involve the white matter of the cerebrum mostly adjacent to the junction with the grey matter, the thalami, the basal ganglia and the brainstem.[1][2] The white matter of the cerebellum and the dentate nuclei are less often involved. However, the brain may appear normal in the neonatal period. The calcifications are visible both with computed tomography and with magnetic resonance imaging.
Magnetic resonance imaging shows additionally diffuse or patchy white matter changes, especially in the periventricular region, the thalami and the internal capsule. Cerebellar and brainstem lesions are less common. Imaging also uncovers parenchymal cysts situated mainly in the thalamic region and more rarely in the brainstem, the parietal lobe and the frontal lobe.
The long bones may be osteopenic and various skeletal changes are found in several patients, such as metaphyseal sclerosis and mild flaring, which is most pronounced in the femur and tibia.[3][4][5]
Laboratory findings
The cerebrospinal fluid and blood tests are typically normal, except for anemia and thrombocytopenia in some children.[1][2][4]
Screening
Because of the rarity of the syndrome, it is not routinely screened before birth. Prenatal diagnosis is possible in families in which a prior child has been diagnosed, because the responsible gene CTC1is known.[7][8]
Management
Seizures are managed with anticonvulsive medications.
Laser coagulation or cryoablation (freezing) of the retina can be used to destroy the abnormal blood vessels. Retinal detachment is repaired with a scleral buckle or with vitrectomy. Removal or enucleation of the eye is a last resort option if the eye already has become blind and painful.
Repeated blood transfusions may be needed to control anemia, and thrombocytopenia can be managed with splenectomy.[1]
Prognosis
The neurological symptoms are progressive and can lead to severe spasticity, bulbar symptoms and dysarthria within one to two decades.[1][3] The life span is shorter than normal. Death occurs between 2 and 30 years of age, depending on the severity of the syndrome. The immediate cause of death is pneumonia, fulminant intestinal bleeding or multiple organ dysfunction.[1]
If not treated, the retinal detachment can lead to ischemia and growth of new blood vessels over the iris and anterior chamber angle. This in turn can cause secondary glaucoma, cataract and, ultimately, blindness of the eye.[1][2]
Epidemiology
This syndrome is usually sporadic although families with two or more affected siblings of both sexes are known.[1][3]
History
A child who was eventually diagnosed with cerebroretinal microangiopathy with calcifications and cysts was first described in the literature in 1987.[9][15] The disorder was suspected of being allelic with either the Revesz syndrome[2] or leukoencephalopathy with calcifications and cysts.[1][2] However, Revesz syndrome, a severe variant of dyskeratosis congenita, was later shown to result from heterozygous dominant mutations in the TINF2 gene,[16] which encodes TRF1-interacting nuclear factor 2, a major component of the telomere protecting shelterin complex, and individuals with the leukoencephalopathy with calcifications and cysts phenotype have not had mutations in the CTC1 gene.[7][8]
Before the causative mutation was identified, mutations were sought from the Wnt signalling pathway because its components were known to be responsible for Norrie disease and familial exudative vitreoretinopathy, which share features of the eye phenotype but not the brain or systemic abnormalities.[1][2]
Coats' disease is named after George Coats.[17][18]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Linnankivi T, Valanne L, Paetau A, Alafuzoff I, Hakumäki JM, Kivelä T, Lönnqvist T, Mäkitie O, Pääkkönen L, Vainionpää L, Vanninen R, Herva R, Pihko H (Oct 2006). "Cerebroretinal microangiopathy with calcifications and cysts". Neurology. 67 (8): 1347–43. doi:10.1212/01.wnl.0000236999.63933.b0. PMID 16943371.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Briggs TA, Abdel-Salam GM, Balicki M, Baxter P, Bertini E, Bishop N, Browne BH, Chitayat D, Chong WK, Eid MM, Halliday W, Hughes I, Klusmann-Koy A, Kurian M, Nischal KK, Rice GI, Stephenson JB, Surtees R, Talbot JF, Tehrani NN, Tolmie JL, Toomes C, van der Knaap MS, Crow YJ (Jan 2008). "Cerebroretinal microangiopathy with calcifications and cysts (CRMCC)". Am J Med Genet A. 146A (2): 182–90. doi:10.1002/ajmg.a.32080. PMID 18076099.
- 1 2 3 4 5 6 7 8 9 10 Crow YJ, McMenamin J, Haenggeli CA, Hadley DM, Tirupathi S, Treacy EP, Zuberi SM, Browne BH, Tolmie JL, Stephenson JB (Feb 2004). "Coats' plus: a progressive familial syndrome of bilateral Coats' disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument". Neuropediatrics. 35 (1): 10–19. doi:10.1055/s-2003-43552. PMID 15002047.
- 1 2 3 4 5 6 Sazgar M, Leonard NJ, Renaud DL, Bhargava R, Sinclair DB (Apr 2002). "Intracranial calcification, retinopathy, and osteopenia: a new syndrome?". Pediatr Neurol. 26 (4): 324–8. doi:10.1016/s0887-8994(01)00398-8. PMID 11992766.
- 1 2 3 Toiviainen-Salo S, Linnankivi T, Saarinen A, Mäyränpää MK, Karikoski R, Mäkitie O (Jun 2011). "Cerebroretinal microangiopathy with calcifications and cysts: characterization of the skeletal phenotype". Am J Med Genet A. 155A (6): 1322–6. doi:10.1002/ajmg.a.33994. PMID 21523908.
- 1 2 Briggs TA, Hubbard M, Hawkins C, Cole T, Livingston JH, Crow YJ, Pigott A (Jan 2011). "Treatment of gastrointestinal bleeding in a probable case of cerebroretinal microangiopathy with calcifications and cysts". Mol Syndromol. 1 (4): 159–62. doi:10.1159/000321559. PMC 3042118
 . PMID 21373254.
. PMID 21373254. - 1 2 3 4 Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafé L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenço CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ (Jan 2012). "Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus". Nat Genet. 44 (3): 338–42. doi:10.1038/ng.1084. PMID 22267198.
- 1 2 3 4 5 Polvi A, Linnankivi T, Kivelä T, Herva R, Keating JP, Mäkitie O, Pareyson D, Vainionpää L, Lahtinen J, Hovatta I, Pihko H, Lehesjoki AE (Mar 2012). "Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts". Am J Hum Genet. 44 (3). doi:10.1016/j.ajhg.2012.02.002.
- 1 2 3 4 Tolmie JL, Browne BH, McGettrick PM, Stephenson JB (1988). "A familial syndrome with Coats' reaction retinal angiomas, hair and nail defects and intracranial calcification". Eye (Lond). 2 (Pt 3): 297–303. doi:10.1038/eye.1988.56. PMID 3402627.
- 1 2 "Entrez Gene: CTC1".
- 1 2 Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R, Leehy K, Heacock M, Price CM, Shippen DE (Oct 2009). "Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes". Mol Cell. 36 (2): 207–18. doi:10.1016/j.molcel.2009.09.017. PMC 2768651. PMID 19854131.
- ↑ "Entrez Gene: OBFC1".
- ↑ "Entrez Gene: TEN1".
- ↑ Chang MM, McLean IW, Merritt JC (Sep 1984). "Coats' disease: a study of 62 histologically confirmed cases". J Pediatr Ophthalmol Strabismus. 21 (5): 163–8. PMID 6502405.
- ↑ McGettrick PM, Loeffler KU (1988). "Bilateral Coats' disease in an infant (a clinical, angiographic, light and electron microscopic study)". Eye (Lond). 1 (Pt 1): 136–45. doi:10.1038/eye.1987.20. PMID 3556653.
- ↑ Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (Feb 2008). "TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita". Am J Hum Genet. 82 (2): 501–9. doi:10.1016/j.ajhg.2007.10.004. PMC 2427222. PMID 18252230.
- ↑ doctor/1926 at Who Named It?
- ↑ Coats G (Nov 1908). "Forms of retinal disease with massive exudation". R Lond Ophthalmol Hosp Rep. 17 (3): 440–525.