DNA nanoball sequencing
DNA nanoball sequencing is a high throughput sequencing technology that is used to determine the entire genomic sequence of an organism. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Fluorescent probes bind to complementary DNA and the probes are then ligated to anchor sequences bound to known sequences on the DNA template. The base order is determined via the fluorescence of the ligated and bound probes.[1]

This DNA sequencing method allows large numbers of DNA nanoballs to be sequenced per run at lower reagent costs compared to other next generation sequencing platforms.[2] However, a limitation of this method is that it generates only short sequences of DNA, which presents challenges to mapping its reads to a reference genome.[1] The company Complete Genomics uses DNA nanoball sequencing to sequence samples submitted by researchers.
Procedure
DNA Nanoball Sequencing involves isolating DNA that is to be sequenced, shearing it into small 400 – 500 base pair (bp) fragments, ligating adapter sequences to the fragments, and circularizing the fragments. The circular fragments are copied by rolling circle replication resulting in many single-stranded copies of each fragment. The DNA copies concatenate head to tail in a long strand, and are compacted into a DNA nanoball. The nanoballs are then adsorbed onto a sequencing flow-cell. Unchained sequencing reactions interrogate specific nucleotide locations in the nanoball by ligating fluorescent probes to the DNA. The color of the fluorescence at each interrogated position is recorded through a high-resolution camera. Bioinformatics are used to analyze the fluorescence data and make a base call, and for mapping the 35-bp mate pair reads to a reference genome. The genome is assembled and any polymorphisms present in the sequence are identified.[1]
DNA Isolation, fragmentation, and size capture

Cells are lysed and DNA is extracted from the cell lysate. The high-molecular-weight DNA, often several megabase pairs long, is sonicated to break the DNA double-strands at random intervals. Bioinformatic mapping of the sequencing reads is most efficient when the sample DNA contains a narrow length range.[3] Therefore, selecting the ideal fragment lengths of the DNA for sequencing the fragments are size separated by polyacrylamide gel electrophoresis (PAGE). The DNA of the suitable size range is purified by gel extraction, resulting in DNA with lengths within a narrow range (typically 400 – 500 base pairs).[1]
Attaching adapter sequences

Adapter DNA sequences must be attached to the unknown DNA fragment so that DNA with known sequences flank the unknown DNA. In the first round of adapter ligation, a right and left adapter (Ad1) is attached to the right and left flanks of the fragmented DNA, and the DNA is PCR amplified (Figure 3). The right and left Ad1 are modified to create complementary single strand ends that bind to each other and form circular DNA. A restriction enzyme is added which cleaves the DNA 13 bp to the right of the right adapter. This results in linear double-stranded DNA (Figure 3). Right and left adapter sequences (Ad2) are ligated onto the ends of the linear DNA and the product is amplified using PCR. The Ad2 sequences are then modified to allow them to bind each other and form circular DNA. The restriction enzyme is used again to cleave the circular DNA 13 bp to the left of Ad1. The result is a linear DNA fragment (Figure 3). Right and left adapter sequences (Ad3) are ligated to the right and left flank of the linear DNA and the product is amplified by PCR. The adapters are modified so that they bind to each other and form circular DNA. The type III restriction enzyme EcoP15 is added, which cleaves the DNA 26 bp to the left of Ad3 and 26 bp to the right of Ad2.[4] This step removes a large segment of DNA and linearizes the DNA once again. Right and left adapters (Ad4) are ligated to the DNA, the product is PCR amplified, and the Ad4 sequences are modified so that they bind to each other. The result is the completed circular DNA template.[1]
Rolling circle replication

Once a circular DNA template, containing sample DNA that is ligated to four unique adapter sequences has been generated, the full sequence is amplified into a long string of DNA. This is accomplished by rolling circle replication with the Phi 29 DNA polymerase which binds and replicates the DNA template (Figure 4). The newly synthesized strand is released from the circular template, resulting in a long single-stranded DNA comprising several head-to-tail copies of the circular template.[5] The four adapter sequences contain palindromic sequences which hybridize and cause the single strand to fold onto itself, resulting in a tight ball of DNA approximately 300 nanometers (nm) across. This allows the nanoballs to remain separated from each other and reduces any tangling between different single stranded DNA lengths.[1]
DNA nanoball microarray

To obtain DNA sequence, the DNA nanoballs are attached to a microarray flow cell (Figure 5). The flow cell is a 25 mm by 75 mm silicon wafer coated with silicon dioxide, titanium, hexamethyldisilazane (HMDS), and a photoresist material. The DNA nanoballs are added to the flow cell and selectively bind to the aminosilane in a highly ordered pattern, allowing a very high density of DNA nanoballs to be sequenced.[1][6]
Unchained sequencing by ligation
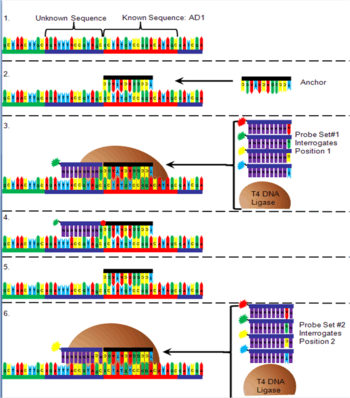
The order of the DNA bases between the adapter sequences is determined after being arrayed onto a flow cell (Figure 6). First, oligonucleotide anchor DNA that is complementary to either the right or left end of one of the adapters is added to the flow cell. Next, T4 DNA ligase is added to a pool of four 10-mer DNA sequences that have degenerate nucleotides in all but one position (for example position 1 next to the anchor, figure) and are added to the flow cell. The interrogative position in the DNA probe contains an "A" nucleotide with a red fluorophore attached, a "C" with a yellow fluorophore attached, a "G" with a green fluorophore attached or a "T" with a blue fluorophore attached. Only the probe that has a complementary nucleotide in the interrogative position will bind. The T4 DNA ligase attaches the probe to the anchor, the non-binding probes are washed away, and the fluorescence is detected. The probe/anchor is removed from the DNA nanoball and another anchor is added. A new pool of probes is added with a different interrogative position. The correct probe hybridizes, is ligated, rinsed and the fluorescence is read and recorded. This process is repeated with all ten interrogation positions next to an anchor sequence. Once all ten positions are recorded, an anchor is added that binds to a different adapter and the process is repeated to identify the ten nucleotides next to that adapter.[1]
Imaging
After each DNA probe/ligation step, the flow cell is imaged to determine which nucleotide base bound to the DNA nanoball. The fluorophore is excited with an arc lamp that radiates specific wavelengths of light towards the flow cell. The wavelength of the fluorescence of each DNA nanoball is captured on a high resolution CCD camera. The image is then processed to remove background noise and assess the intensity of each point. The color of each DNA nanoball corresponds to a base at the interrogative position and a computer records the base position information. The order of 70 nucleotides per DNA nanoball is determined using 80 images (8 rounds of 10 interrogative positions per round).[1]
Genome assembly
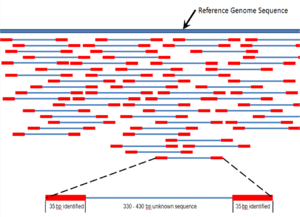
In generating the circular template, a large segment of the original 400 – 500 base pair fragment was replaced with the adapter Ad4. The 70 bp that are sequenced are therefore the first 35 bp of the original 400 – 500 bp fragment, and the last 35 bp of the 400 – 500 bp fragment. Therefore, the sequence is identified for two 35 bp reads of DNA separated by about 330 – 430 bp. These 35 bp reads are compared, using bioinformatics, to a reference genome and assigned to a genetic locus (Figure 7).[7] Massive parallel genome mapping is accomplished through high coverage of the reference nucleotide positions, and the complete genome of the DNA sample is assembled. Single base-pair mismatches between the sequenced reads and the reference sequence are used to identify possible single nucleotide polymorphism (SNP). In addition, mapping of one 35-bp portion of the mate pair may identify DNA inserts and deletions (indels) via bioinformatics algorithms that detect possible discrepancies between the mate pairs.[1]
Advantages

DNA nanoball sequencing technology offers several advantages over other sequencing platforms. One major advantage is the use of very high-density arrays. The array design permits one DNA nanoball to attach to each pit that is part of an ordered array, and therefore a higher concentration of DNA can be added. This allows a high percentage of the pits to be occupied by a DNA nanoball thus maximizing the number of reads per flow cell (Figure Top).[1] compared to other sequencing arrays where molecules of DNA are added to a flow cell in a random orientation (Figure bottom)[8]
Another important advantage is that the sequencing reactions are non-progressive; after each reading of the probe, the probe and anchor are removed and a new anchor and probe set are added. Therefore, if a probe did not bind in the previous reaction, this has no effect on the next probe ligation,[2] thus eliminating a major source of reading error that may occur in other next generation sequencing platforms.[2] It also reduces the use of expensive probes, since DNA nanoball sequencing does not necessitate the probe ligation reaction to be run to completion.[2]
Other advantages of DNA nanoball sequencing include the use of high-fidelity Phi 29 DNA polymerase[5] to ensure accurate amplification of the circular template, several hundred copies of the circular template compacted into a small area resulting in an intense signal, and attachment of the fluorophore to the probe at a long distance from the ligation point results in improved ligation.[1]
Disadvantages
The main disadvantage of DNA nanoball sequencing is the short read length of the DNA sequences obtained with this method.[1] Short reads, especially for DNA high in DNA repeats, may map to two or more regions of the reference genome. A second disadvantage of this method is that multiple rounds of PCR have to be used. This can introduce PCR bias and possibly amplify contaminants in the template construction phase.[1]
Applications
DNA nanoball sequencing has been used in recent studies. Lee et al. used this technology to find mutations that were present in a lung cancer and compared them to normal lung tissue.[9] They were able to identify over 50,000 single nucleotide variants Roach et al. used DNA nanoball sequencing to sequence the genomes of a family of four relatives and were able to identify SNPs that may be responsible for a Mendelian disorder,[10] and were able to estimate the inter-generation mutation rate.[10] The Institute for Systems Biology has used this technology to sequence 615 complete human genome samples as part of a survey studying neurodegenerative diseases, and the National Cancer Institute is using DNA nanoball sequencing to sequence 50 tumours and matched normal tissues from pediatric cancers.
Significance
Massively parallel next generation sequencing platforms like DNA nanoball sequencing may contribute to the diagnosis and treatment of many genetic diseases. The cost of sequencing an entire human genome has fallen from about one million dollars in 2008, to $4400 in 2010 with the DNA nanoball technology.[11] Sequencing the entire genomes of patients with heritable diseases or cancer, mutations associated with these diseases have been identified, opening up strategies, such as targeted therapeutics for at-risk people and for genetic counseling.[11] As the price of sequencing an entire human genome approaches the $1000 mark, genomic sequencing of every individual may become feasible as part of normal preventative medicine.[11]
Notes
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Drmanac, R.; Sparks, A. B.; Callow, M. J.; Halpern, A. L.; Burns, N. L.; Kermani, B. G.; Carnevali, P.; Nazarenko, I.; et al. (2009). "Human Genome Sequencing Using Unchained Base Reads on Self-Assembling DNA Nanoarrays". Science. 327 (5961): 78–81. doi:10.1126/science.1181498. PMID 19892942.
- 1 2 3 4 Porreca, Gregory J (2010). "Genome sequencing on nanoballs". Nature Biotechnology. 28 (1): 43–4. doi:10.1038/nbt0110-43. PMID 20062041.
- ↑ Fullwood, M. J.; Wei, C.-L.; Liu, E. T.; Ruan, Y. (2009). "Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses". Genome Research. 19 (4): 521–32. doi:10.1101/gr.074906.107. PMID 19339662.
- ↑ "Figure 3". from Dryden, D. T. F.; Murray, NE; Rao, DN (2001). "Nucleoside triphosphate-dependent restriction enzymes". Nucleic Acids Research. 29 (18): 3728–41. doi:10.1093/nar/29.18.3728. PMC 55918
 . PMID 11557806.
. PMID 11557806. - 1 2 Blanco, Luis; Bernad, Antonio; Lázaro, José M.; Martin, Gil; Garmendia, Cristina; Margarita, M; Salas (1989). "Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication". The Journal of Biological Chemistry. 264 (15): 8935–40. PMID 2498321.
- ↑ Chrisey, L.; Lee, GU; O'Ferrall, CE (1996). "Covalent attachment of synthetic DNA to self-assembled monolayer films". Nucleic Acids Research. 24 (15): 3031–9. doi:10.1093/nar/24.15.3031. PMC 146042. PMID 8760890.
- ↑ Li, Yingrui; Hu, Yujie; Bolund, Lars; Wang, Jun (2010). "State of the art de novo assembly of human genomes from massively parallel sequencing data". Human genomics. 4 (4): 271–7. doi:10.1186/1479-7364-4-4-271. PMID 20511140.
- ↑ Bentley, David R.; Balasubramanian, Shankar; Swerdlow, Harold P.; Smith, Geoffrey P.; Milton, John; Brown, Clive G.; Hall, Kevin P.; Evers, Dirk J.; et al. (2008). "Accurate whole human genome sequencing using reversible terminator chemistry". Nature. 456 (7218): 53–9. doi:10.1038/nature07517. PMC 2581791. PMID 18987734.
- ↑ Lee, William; Jiang, Zhaoshi; Liu, Jinfeng; Haverty, Peter M.; Guan, Yinghui; Stinson, Jeremy; Yue, Peng; Zhang, Yan; et al. (2010). "The mutation spectrum revealed by paired genome sequences from a lung cancer patient". Nature. 465 (7297): 473–7. doi:10.1038/nature09004. PMID 20505728.
- 1 2 Roach, J. C.; Glusman, G.; Smit, A. F. A.; Huff, C. D.; Hubley, R.; Shannon, P. T.; Rowen, L.; Pant, K. P.; et al. (2010). "Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing". Science. 328 (5978): 636–9. doi:10.1126/science.1186802. PMC 3037280. PMID 20220176.
- 1 2 3 Speicher, Michael R; Geigl, Jochen B; Tomlinson, Ian P (2010). "Effect of genome-wide association studies, direct-to-consumer genetic testing, and high-speed sequencing technologies on predictive genetic counselling for cancer risk". The Lancet Oncology. 11 (9): 890–8. doi:10.1016/S1470-2045(09)70359-6. PMID 20537948.