HLA-A
| MHC class I, A (heterodimer) | ||
 | ||
| Illustration of HLA-A | ||
| − | ||
| Protein type | Cell surface receptor | |
| Function | Peptide presentation for immune recognition | |
| − | ||
| Subunit name | Gene | Chromosomal locus |
| α | HLA-A | Chromosome 6p21.3 |
| β2M | B2M | Chromosome 15q22 |
HLA-A is a group of human leukocyte antigens (HLA) that are coded for by the HLA-A locus, which is located at human chromosome 6p21.3.[1] HLA is a major histocompatibility complex (MHC) antigen specific to humans. HLA-A is one of three major types of human MHC class I cell surface receptors. The others are HLA-B and HLA-C.[2] The receptor is a heterodimer, and is composed of a heavy α chain and smaller β chain. The α chain is encoded by a variant HLA-A gene, and the β chain (β2-microglobulin) is an invariant β2 microglobulin molecule.[3] The β2 microglobulin protein is coded for by a separate region of the human genome.[4]
MHC Class I molecules such as HLA-A are part of a process that presents short polypeptides to the immune system. These polypeptides are typically 7-11 amino acids in length and originate from proteins being expressed by the cell. There are two classes of polypeptide that can be presented by an HLA protein: those that are supposed to be expressed by the cell (self) and those of foreign derivation (non-self).[5] Under normal conditions cytotoxic T cells, which normally patrol the body in the blood, "read" the peptide presented by the complex. T cells, if functioning properly, only bind to non-self peptides. If binding occurs, a series of events is initiated culminating in cell death via apoptosis.[6] In this manner, the human body eliminates any cells infected by a virus or expressing proteins they shouldn't be (e.g. cancerous cells).
For humans, as in most mammalian populations, MHC Class I molecules are extremely variable in their primary structure, and HLA-A is ranked among the genes in humans with the fastest-evolving coding sequence. As of December 2013, there are 2432 known HLA-A alleles coding for 1740 active proteins and 117 null proteins.[2] This level of variation on MHC Class I is the primary cause of transplant rejection, as random transplantation between donor and host is unlikely to result in a matching of HLA-A, B or C antigens. Evolutionary biologists also believe that the wide variation in HLAs is a result of a balancing act between conflicting pathogenic pressures. Greater variety of HLAs decreases the probability that the entire population will be wiped out by a single pathogen as certain individuals will be highly resistant to each pathogen.[5] The effect of HLA-A variation on HIV/AIDS progression is discussed below.
HLA-A gene
| View/Edit Human | View/Edit Mouse |
The HLA-A gene is located on the short arm of chromosome 6 and encodes the larger, α-chain, constituent of HLA-A. Variation of HLA-A α-chain is key to HLA function. This variation promotes genetic diversity in the population. Since each HLA has a different affinity for peptides of certain structures, greater variety of HLAs means greater variety of antigens to be 'presented' on the cell surface, enhancing the likelihood that a subset of the population will be resistant to a given foreign invader. This decreases the likelihood that a single pathogen has the capability to wipe out the entire human population.
Each individual can express up to two types of HLA-A, one from each of their parents. Some individuals will inherit the same HLA-A from both parents, decreasing their individual HLA diversity; however, the majority of individuals will receive two different copies of HLA-A. This same pattern follows for all HLA groups.[9] In other words, every single person can only express either one or two of the 2432 known HLA-A alleles.
Alleles
All HLAs are assigned a name by the World Health Organization Naming Committee for Factors of the HLA System. This name is organized to provide the most information about the particular allele while keeping the name as short as possible. An HLA name looks something like this:
HLA-A*02:01:01:02L
All alleles receive at least a four digit classification (HLA-A*02:12). The A signifies which HLA gene the allele belongs to. There are many HLA-A alleles, so that classification by serotype simplifies categorization. The next pair of digits indicates this assignment. For example, HLA-A*02:02, HLA-A*02:04, and HLA-A*02:324 are all members of the A2 serotype (designated by the *02 prefix).[2] This group is the primary factor responsible for HLA compatibility. All numbers after this cannot be determined by serotyping and are designated through gene sequencing. The second set of digits indicates what HLA protein is produced. These are assigned in order of discovery and as of December 2013 there are 456 different HLA-A*02 proteins known (assigned names HLA-A*02:01 to HLA-A*02:456). The shortest possible HLA name includes both of these details.[1] Each extension beyond that signifies a nucleotide change that may or may not change the protein and the interpretation of the extensions is covered in greater detail in current HLA naming system.
Protein
The protein coded for by the HLA-A gene is 365 amino acids long and weighs roughly 41,000 Daltons (Da).[10] It contains 8 exons.[11]
| Exon | Protein segment |
|---|---|
| 1 | Signal peptide |
| 2 | α1 domain |
| 3 | α2 domain |
| 4 | α3 domain |
| 5 | transmembrane region |
| 6 | cytoplasmic tail |
| 7 | cytoplasmic tail |
| 8 | Unspecified |
The HLA-A signal peptide is a series of hydrophobic amino acids present at the N-terminus of the protein that directs it to the endoplasmic reticulum where the remaining seven domains are translated.[10][11][12] The three α domains form the binding groove that holds a peptide for presentation to CD8+ t-cells. The transmembrane region is the region that is embedded in the phospholipid bilayer surrounding the ER lumen.[11] The HLA-A protein is a single-pass transmembrane protein.[10] In other words, the first four domains of the protein are inside the ER lumen, while the last three domains are present outside the lumen, giving the protein the orientation required for proper function. The last three domains of the protein form a tail of primarily β-sheets that remains in the cell's cytosol.[11]
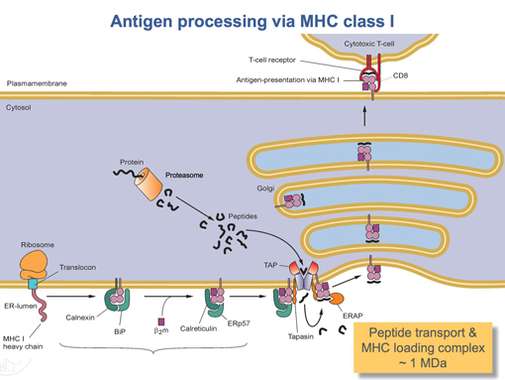
Once the HLA-A protein is completely translated, it must be folded into the proper shape. A molecular chaperone protein called calnexin and an enzyme called ERp57 assist in the folding process. Calnexin holds the HLA-A heavy chain while Erp57 catalyzes disulfide bonds between the heavy chain and the light, β2-microglobulin chain. This bond induces a conformational change in the heavy chain, forming the binding groove. Calnexin then dissociates with the complex, now referred to as a peptide loading complex, and is replaced by calreticulin, another chaperone protein. Short peptides are continually transported from around the cell into the ER lumen by a specialized transport protein called TAP. TAP then binds to the peptide loading complex along with another protein, called tapasin. At this point the peptide loading complex consists of HLA-A (heavy chain), β2-microglobulin (light chain), an ERp57 enzyme, calreticulin chaperone protein, TAP (with a bound peptide fragment), and tapasin. Tapasin increases the stability of TAP, in addition to stabilizing the entire peptide loading complex. At this point TAP releases the peptide it transported into the ER lumen. The proximity of the HLA-A binding groove to TAP is ensured by the peptide loading complex. This increases the likelihood that the peptide will find the groove. If the peptide's affinity for the HLA-A protein is great enough, it binds in the groove.[14] Research suggests that tapasin may actively load peptides from TAP into the HLA-A complex while also holding class I molecules in the ER lumen until a high affinity peptide has been bound.[15]
After a peptide of high enough affinity has bonded to the class I MHC, calreticulin, ERp57, TAP, and tapasin release the molecule.[14] At this point the class I complex consists of an HLA-A protein bonded to a β2-microglobulin and a short peptide. It is still anchored in the ER membrane by the transmembrane domain. At some point the ER will receive a signal and the portion of the membrane holding the complex will bud off and be transported to the golgi bodies for further processing. From the golgi bodies, the complex is transported, again via vesicle transport, to the cell membrane. This is the point at which the orientation mentioned previously becomes important. The portion of the HLA-A complex holding the peptide must be on the exterior surface of the cell membrane. This is accomplished by vesicle fusion with the cell membrane.[12]
Function
Natural function
MHC Class I molecules present small peptides, typically 7-10 amino acids in length, to the immune system. A glycoprotein called CD8 binds to residues 223-229 in the α3 domain of HLA-A and this glycoprotein stabilizes interactions between the t-cell receptor on cytotoxic (CD8+) T-lymphocytes and the Class I MHC.[16] The T-cell receptor also has the potential to bind to the peptide being presented by the MHC. In a properly functioning immune system, only t-cells that do not bind self peptides are allowed out of the thymus, thus, if a T-cell binds to the peptide, it must be a foreign or abnormal peptide. The t-cell then initiates apoptosis, or programmed cell death. This process can happen as quickly as 5 minutes after initial foreign antigen presentation, although typically it takes several hours for death to become apparent.[17] This process is the basis of acquired immunity and serves as the primary defense against viruses and other intracellular pathogens.
Other activities
By the 1960s, it became evident that factors on donated organs and tissues often resulted in destruction of the donated tissue by the host's immune system. MHCs were originally discovered as a result of this observation (see history of HLAs for more details).[5] There are two types of peptide presenting complexes, Class I and Class II MHCs. Each of these has multiple HLA genes, of which HLA-A is but one. There are three major HLAs that should be matched between donors and recipients. They are HLA-A, HLA-B, (both Class I MHCs) and HLA-DR (a Class II MHC).[9] If the two tissues have the same genes coding for these three HLAs, the likelihood and severity of rejection is minimized.[18]
Role in disease
| Associated disease | Serotypes | ||
| Ankylosing spondylitis | A24 | ||
| Diabetes, type-I[19] | A1 | A24 | |
| Hemochromatosis (lower CD8+ cells) | A3 | ||
| Myasthenia gravis | A3 | A24 | A30 |
| Leukemia, T-cell, Adult | A26 | A68 | |
| Multiple sclerosis | A3 | ||
| Papilloma virus susept. | A11 | ||
| Spontaneous abortion | A2 | ||
HLAs serve as the sole link between the immune system and what happens inside cells. Thus any alteration on the part of the HLA, be it decreased binding to a certain peptide or increased binding to a certain peptide, is expressed as, respectively, increased susceptibility to disease or decreased susceptibility to disease. In other words, certain HLAs may be incapable of binding any of the short peptides produced by proteolysis of pathogenic proteins. If this is the case, there is no way for the immune system to tell that a cell is infected. Thus the infection can proliferate largely unchecked. It works the other way too. Some HLAs bind pathogenic peptide fragments with very high affinity. This in essence "supercharges" their immune system in regards to that particular pathogen, allowing them to manage an infection that might otherwise be devastating.[5]
HIV/AIDS
One of the most researched examples of differential immune regulation of a pathogen is that of human immunodeficiency virus. Because HIV is an RNA virus, it mutates incredibly quickly. This changes the peptides produced via proteolysis, which changes the peptides able to be presented to the immune system by the infected cell's MHCs. Any virus with a mutation that creates a peptide with high affinity for a particular HLA is quickly killed by the immune system, and thus does not survive and that high affinity peptide is no longer produced. However, it turns out that even HIV has some conserved regions in its genome, and if a HLA is capable of binding to a peptide produced from a conserved region, there is little the HIV can do to avoid immune detection and destruction.[5] This is the principle behind HLA-mediated differential HIV loads.
With over 2000 variations of the HLA-A coded MHC, it is difficult to determine the impact of all variants upon HIV loads. However, a select few have been implicated. HLA-A*30 has been shown to decrease viral load to less than 10,000 copies/cubic millimeter, considered quite low. On the other hand, HLA-A*02 has been implicated in high viral load (greater than 100,000 copies/cubic millimeter) when associated with HLA-B*45. Additionally, the haplotypes HLA-A*23-C*07 and HLA-A*02-C*16 typically expressed increased viral loads within the sample population of Zambians. One of the most effective HIV-inhibiting haplotypes was HLA-A*30-C*03 while one of the least effective was HLA-A*23*B*14. In summation, HLA-A*23 was highly correlated with increased HIV load among the sample population, although it is important to note that across samples of differing ethnicity this correlation decreases significantly.[20]
Although classification of the effect of individual HLA genes and alleles on the presence of HIV is difficult, there are still some strong conclusions that can be made. Individuals who are homozygous in one or more Class I HLA genes typically progress to AIDS much more rapidly than heterozygotes. In some homozygous individuals the rate of progression is double that of heterozygotes. This differential progression is correlated fairly tightly with the degree of heterozygosity.[21] In summation, certain HLA-A alleles are associated with differing viral loads in HIV infected patients; however, due to the diversity amongst those alleles, it is difficult to classify each and every allele's impact upon immune regulation of HIV. Nevertheless, it is possible to correlate heterozygosity in HLA-A alleles to decreased rate of progression to AIDS.
Not only do certain HLA alleles prescribe increased or decreased resistance to HIV, but HIV is able to alter HLA expression, and does so selectively to avoid being eliminated by natural killer cells (NK cells). Research has shown that HIV downregulates Class I MHC expression in infected cells. However, doing so indiscriminately opens up the opportunity for attack by NK cells, because NK cells respond to downregulation of HLA-C and HLA-E. Obviously, this mechanism has put selective pressure on the HIV virus. Thus, HIV has evolved the capability to downregulate HLA-A and HLA-B without significantly disturbing the expression of HLA-C and HLA-E.[22] A protein coded for by the HIV genome, negative regulatory factor (Nef), induces this change by binding to the cytoplasmic tail of the Class I MHC while it is still in the endoplasmic reticulum or occasionally while it is in the early stages of trafficking through the golgi bodies. This complex of MHC and Nef then causes adaptor protein 1 (AP-1) to direct the MHC to the lysosomes for degradation instead of to the cell membrane where it normally functions.[23] In addition to selective HLA downregulation, negative regulatory factor (Nef) enables HIV to downregulate CD4 and CD8. These glycoproteins are essential for, respectively, helper t-cell and cytotoxic t-cell binding to MHCs. Without these cofactors, both types of t-cells are less likely to bind to HLAs and initiate apoptosis, even if the HLA is expressing an HIV derived (non-self) peptide. Both of these proteins are also targeted at their cytoplasmic tail domain.[23] The combination of these abilities greatly enhances HIV's ability to avoid detection by the immune system.
Summary
HLA-A is one particular group of the human Class I MHCs. It consists of several hundred different genes and several thousand variant alleles. HLA-A is critical to the cytotoxic t-cell controlled immune response to viruses and other intracellular pathogens. Because each HLA-A gene has a high affinity for slightly different peptides, certain HLA-As are associated with increased risk, more rapid progression, and/or increased severity of many diseases. For similar reasons, HLA-A matching is essential to successful tissue transplants.
References
- 1 2 "HLA Nomenclature @ hla.alleles.org". Anthony Nolan Research Institute. 10 Nov 2013. Retrieved 8 Dec 2013.
- 1 2 3 "Statistics". European Bioinformatics Institute (EBI) / European Molecular Biology Laboratory (EMBL). Retrieved 13 Dec 2013.
- ↑ Delves PJ (Aug 2013). "Human Leukocyte Antigen (HLA) System: Biology of the Immune System". Merck Manual Professional. Merck Sharp & Dohme Corp. Retrieved 14 Dec 2013.
- ↑ "B2M Gene". GeneCards. Weizmann Institute of Science. 7 Nov 2013. Retrieved 14 Dec 2013.
- 1 2 3 4 5 Daniel M. Davis (2014). The Compatibility Gene. How Our Bodies Fight Disease, Attract Others, and Define Our Selves. Oxford: Oxford University Press. ISBN 0-19-931641-4.
- ↑ Accorsi D (14 Sep 2012). "MHC class I assembly and presentation". YouTube. Retrieved 8 Dec 2013.
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- 1 2 Fix M (1998). "HLA Matching, Antibodies, and You". Kidney Transplantation: Past, Present, and Future. University of Michigan Medical Center/Stanford University. Retrieved 14 Dec 2013.
- 1 2 3 "Major Histocompatibility Complex, Class I, A". Gene Cards. Weizmann Institute of Science. 7 November 2013. Retrieved 16 December 2013.
- 1 2 3 4 "HLA-A major histocompatibility complex, class I, A [Homo sapiens (human)]". National Center for Biotechnology Information. U.S. National Library of Medicine. 12 December 2013. Retrieved 16 December 2013.
- 1 2 Alberts, Bruce (2010). Essential Cell Biology (Third ed.). Garland Science. ISBN 9780815341291.
- ↑ Tampé, Robert. "P16 Translocation mechanism and viral inhibition of the MHC I peptide-loading complex". Molecular Understanding of Transmembrane Processes. Institute of Biochemistry Biocenter. Retrieved 16 December 2013.
- 1 2 Accorsi, Diego (14 September 2012). "MHC class I assembly and presentation". Immunology Toronto. Department of Immunology and Biochemistry and Biomedical Communications at the University of Toronto. Retrieved 16 December 2013.
- ↑ Grandea AG, Van Kaer L (April 2001). "Tapasin: an ER chaperone that controls MHC class I assembly with peptide". Trends in Immunology. 22 (4): 194–9. doi:10.1016/S1471-4906(01)01861-0. PMID 11274924.
- ↑ "CD8". T-cell Modulation Group. tcells.org. 2009. Retrieved 17 December 2013.
- ↑ Janeway, Charles A. (2001). "8". Immunobiology the immune system health & disease (5. ed.). New York: Garland. ISBN 081533642X. Retrieved 17 December 2013.
- ↑ Solomon S, Pitossi F, Rao MS (February 2015). "Banking on iPSC--is it doable and is it worthwhile". Stem Cell Reviews. 11 (1): 1–10. doi:10.1007/s12015-014-9574-4. PMC 4333229
 . PMID 25516409.
. PMID 25516409. - ↑ Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA (August 2002). "The HLA class I A locus affects susceptibility to type 1 diabetes". Human Immunology. 63 (8): 657–64. doi:10.1016/S0198-8859(02)00421-4. PMID 12121673.
- ↑ Tang J, Tang S, Lobashevsky E, Myracle AD, Fideli U, Aldrovandi G, Allen S, Musonda R, Kaslow RA (August 2002). "Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1". Journal of Virology. 76 (16): 8276–84. doi:10.1128/JVI.76.16.8276-8284.2002. PMC 155130. PMID 12134033.
- ↑ Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O'Brien SJ (March 1999). "HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage". Science. 283 (5408): 1748–52. doi:10.1126/science.283.5408.1748. PMID 10073943.
- ↑ Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D (June 1999). "The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells". Immunity. 10 (6): 661–71. doi:10.1016/S1074-7613(00)80065-5. PMID 10403641.
- 1 2 Leonard JA, Filzen T, Carter CC, Schaefer M, Collins KL (July 2011). "HIV-1 Nef disrupts intracellular trafficking of major histocompatibility complex class I, CD4, CD8, and CD28 by distinct pathways that share common elements". Journal of Virology. 85 (14): 6867–81. doi:10.1128/JVI.00229-11. PMC 3126561. PMID 21543478.
External links
- Molecular Anthropology Yahoo Group
- HLA Allele and Haplotype Frequency Database
- HLA Nomenclature Full List of Class I Proteins
PDB gallery | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||