Sucrose intolerance
| Sucrose intolerance | |
|---|---|
 | |
| Sucrose | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E74.3 |
| ICD-9-CM | 271.3 |
| OMIM | 222900 |
| DiseasesDB | 29844 |
Sucrose intolerance, also called congenital sucrase-isomaltase deficiency (CSID)[1] or sucrase-isomaltase deficiency,[2] Genetic sucrase-isomaltase deficiency (GSID) or sucrase-isomaltase deficiency, is the condition in which sucrase-isomaltase, an enzyme needed for proper metabolism of sucrose (sugar) and starch (i.e., grains and rice), is not produced or the enzyme produced is either partially functional or non-functional in the small intestine. All GSID patients lack fully functional sucrase, while the isomaltase activity can vary from minimal functionality to almost normal activity. The presence of residual isomaltase activity may explain why some GSID patients are better able to tolerate starch in their diet than others with GSID. The highest prevalence rates are seen in the Inuit populations of Greenland (5%-10%), Alaska (3%-7%) and Canada (about 3%). European descent prevalence ranges from 0.2% to 0.05%. There is a lower prevalence reported in African Americans and Hispanics compared to Caucasians.[3] [4]
Overview
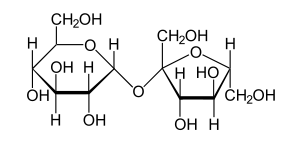
Sucrose (also termed saccharose) is a disaccharide and is a two-sugar chain composed of glucose and fructose which are bonded together. A more familiar name is table, beet, or cane sugar. It was believed that most cases of sucrose intolerance were to do an autosomal recessive, genetic, metabolic disease. Based on new data patients with heterozygous and compound heterozygous genotypes can have symptom presentation as well. GSID involves deficiency in the enzyme sucrase-isomaltase, which breaks apart the glucose and fructose molecules. When disaccharides are consumed, they must be broken down into monosaccharides by enzymes in the intestines before they can be absorbed. Monosaccharides, or single sugar units, are absorbed directly into the blood.[5]
A deficiency of sucrase may result in malabsorption of sugar, which can lead to potentially serious symptoms. Since sucrose-isomaltase is involved in the digestion of starches, some GSID patients may not be able to absorb starches as well. It is important for those with sucrose intolerance to minimize sucrose consumption as much as possible. Dietary supplements or medications may be taken as a substitute for the enzyme missing or to introduce healthy bacteria into the immune system.
Sucrose intolerance can be caused by genetic mutations in which both parents must contain this gene for the child to carry the disease (so-called primary sucrose intolerance). Sucrose intolerance can also be caused by irritable bowel syndrome, aging, or small intestine disease (secondary sucrose intolerance). There are specific tests used to help determine if a person has sucrose intolerance. The most accurate test is the enzyme activity determination, which is done by biopsying the small intestine. This test is a diagnostic for GSID. Other tests which can aid in the diagnosis of GSID but which are not truly diagnostic for the disease are the sucrose breath test, and a genetic test which tests for the absence of certain genes which are thought to be responsible for GSID.[6]
Signs and symptoms
- Abdominal cramps and bloating
- Diarrhea and constipation
- Vomiting
- Hypoglycemia and headaches
- Poor weight gain and growth
- Upper respiratory tract and viral diseases
- Anxiety and heart palpitations
- Excess gas production
See also
References
- ↑ Sander P, Alfalah M, Keiser M, et al. (January 2006). "Novel mutations in the human sucrase-isomaltase (SI) gene that cause congenital carbohydrate malabsorption". Hum. Mutat. 27 (1): 119. doi:10.1002/humu.9392. PMID 16329100.
- ↑ Baudon JJ, Veinberg F, Thioulouse E, Morgant G, Aymard P, Charritat JL (April 1996). "Sucrase-isomaltase deficiency: changing pattern over two decades". J. Pediatr. Gastroenterol. Nutr. 22 (3): 284–8. doi:10.1097/00005176-199604000-00010. PMID 8708882.
- ↑ Treem, William R. (2012). "Clinical Aspects and Treatment of Congenital Sucrase-Isomaltase Deficiency". Journal of Pediatric Gastroenterology and Nutrition. 55: S7–S13. doi:10.1097/01.mpg.0000421401.57633.90.
- ↑ Uhrich, Stefanie; Wu, Zaining; Huang, Jie-Yu; Scott, C. Ronald (2012). "Four Mutations in the SI Gene Are Responsible for the Majority of Clinical Symptoms of CSID". Journal of Pediatric Gastroenterology and Nutrition. 55: S34–S35. doi:10.1097/01.mpg.0000421408.65257.b5.
- ↑ Treem, William R. (2012). "Clinical Aspects and Treatment of Congenital Sucrase-Isomaltase Deficiency". Journal of Pediatric Gastroenterology and Nutrition. 55: S7–S13. doi:10.1097/01.mpg.0000421401.57633.90.
- ↑ Uhrich, Stefanie; Wu, Zaining; Huang, Jie-Yu; Scott, C. Ronald (2012). "Four Mutations in the SI Gene Are Responsible for the Majority of Clinical Symptoms of CSID". Journal of Pediatric Gastroenterology and Nutrition. 55: S34–S35. doi:10.1097/01.mpg.0000421408.65257.b5.