Jackson–Weiss syndrome
| Jackson–Weiss syndrome | |
|---|---|
 | |
|
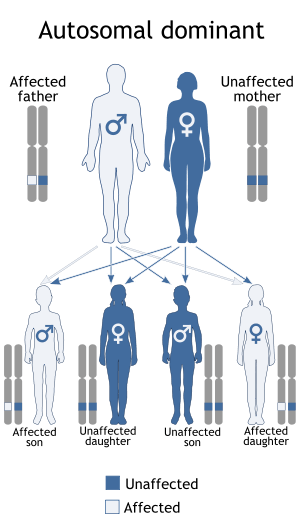
Jackson–Weiss syndrome is inherited in an autosomal dominant pattern. DiseasesDB = 31364 | |
| Classification and external resources | |
| OMIM | 123150 |
Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face.[1] It can also sometimes cause intellectual disability and crossed eyes as well.
It was characterized in 1976.[2]
Presentation
Many of the characteristic facial features of Jackson–Weiss syndrome result from the premature fusion of the skull bones and foot bones. The head is unable to grow normally, which can lead to a misshapen skull, widely spaced eyes, and a bulging forehead. Foot abnormalities are the most consistent characteristic, as not all individuals with Jackson–Weiss syndrome have abnormal skull or facial features. The big toes are enlarged and bend away from the other toes. Hand abnormalities are rare. People with Jackson–Weiss syndrome usually have normal intelligence and a normal life span.
Genetics
Mutations in the FGFR2 gene cause Jackson–Weiss syndrome.[3] The FGFR2 gene produces a protein called fibroblast growth factor receptor 2.[4] It occurs in chromosome number 10. Among its multiple functions, this protein signals immature cells to become bone cells in a developing embryo and fetus. A mutation in a specific part of the FGFR2 gene alters the protein and causes prolonged signaling, which promotes the premature fusion of bones in the skull and feet.
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Treatment
Treatment can be done through surgery (multi-staged) for some facial features, feet, and fingers.[5]
Epidemiology
Jackson–Weiss syndrome is a rare genetic disorder; its incidence is unknown.
References
- ↑ Heike C, Seto M, Hing A, Palidin A, Hu FZ, Preston RA, Ehrlich GD, Cunningham M (2001). "Century of Jackson–Weiss syndrome: further definition of clinical and radiographic findings in "lost" descendants of the original kindred". Am J Med Genet. 100 (4): 315–24. doi:10.1002/ajmg.1266. PMID 11343323.
- ↑ Jackson CE, Weiss L, Reynolds WA, Forman TF, Peterson JA (June 1976). "Craniosynostosis, midfacial hypoplasia and foot abnormalities: an autosomal dominant phenotype in a large Amish kindred". J. Pediatr. 88 (6): 963–8. doi:10.1016/S0022-3476(76)81050-5. PMID 1271196.
- ↑ Jabs EW, Li X, Scott AF, Meyers G, Chen W, Eccles M, Mao JI, Charnas LR, Jackson CE, Jaye M (1994). "Jackson–Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2". Nat Genet. 8 (3): 275–9. doi:10.1038/ng1194-275. PMID 7874170.
- ↑ Chen L, Deng CX (2005). "Roles of FGF signaling in skeletal development and human genetic diseases". Front Biosci. 10 (1-3): 1961–76. doi:10.2741/1671. PMID 15769677.
- ↑ Fryns, Buggenhout, Jean, Griet (July 2005). "Jackson–Weiss syndrome" (PDF). p. 2. Retrieved 2009-03-31.