Mascot (software)
| Original author(s) | David Perkins and Darryl Pappin |
|---|---|
| Initial release | 1999 |
| Stable release |
2.4.01
/ October 2012 |
| Development status | Active |
| Operating system | Linux or Windows |
| Available in | C |
| Type | Protein identification Bioinformatics |
| License | proprietary, free for online use |
| Website | http://www.matrixscience.com/ |
Mascot is a software search engine that uses mass spectrometry data to identify proteins from peptide sequence databases.[1][2] Mascot is widely used by research facilities around the world. Mascot uses a probabilistic scoring algorithm for protein identification that was adapted from the MOWSE algorithm. Mascot is freely available to use on the website of Matrix Science . A License is required for in-house use where more features can be incorporated.
History
MOWSE was one of the first algorithms developed for protein identification using peptide mass fingerprinting.[3] It was originally developed in 1993 as a collaboration between Darryl Pappin of the Imperial Cancer Research Fund (ICRF) and Alan Bleasby of the Science and Engineering Research Council (SERC). MOWSE stood apart from other protein identification algorithms in that it produced a probability-based score for identification. It was also the first to take into account the non-uniform distribution of peptide sizes, caused by the enzymatic digestion of a protein that is needed for mass spectrometry analysis. However, MOWSE was only applicable to peptide mass fingerprint searches and was dependent on pre-compiled databases which were inflexible with regard to post-translational modifications and enzymes other than trypsin. To overcome these limitations, to take advantage of multi-processor systems and to add non-enzymatic search functionality, development was begun again from scratch by David Perkins at the Imperial Cancer Research Fund. The first versions were developed for Silicon Graphics Irix and Digital Unix systems. Eventually this software was named Mascot and to reach a wider audience, an external bioinformatics company named Matrix Science was created by David Creasey and John Cotterel to develop and distribute Mascot. Legacy software versions exist for Tru64, Irix, AIX, Solaris, Microsoft Windows NT4 and Microsoft Windows 2000. Mascot has been available as a free and unrestricted service on the Matrix Science website since 1999 and has been cited in scientific literature over 5,000 times. Matrix Science still continues to work on improving Mascot’s functionality.
Applications
Mascot identifies proteins by interpreting mass spectrometry data. The prevailing experimental method for protein identification is a bottom-up approach, where a protein sample is typically digested with Trypsin to form smaller peptides. While most proteins are too big, peptides usually fall within the limited mass range that a typical mass spectrometer can measure. Mass spectrometers measure the molecular weights of peptides in a sample. Mascot then compares these molecular weights against a database of known peptides. The program cleaves every protein in the specified search database in silico according to specific rules depending on the cleavage enzyme used for digestion and calculates the theoretical mass for each peptide. Mascot then computes a score based on the probability that the peptides from a sample match those in the selected protein database. The more peptides Mascot identifies from a particular protein, the higher the Mascot score for that protein.
Features
- Peptide Mass Fingerprint search
- Identifies proteins from an uploaded peak list using a technique known as peptide mass fingerprinting.
- Sequence query
- Combines peptide mass data with amino acid sequence and composition information usually obtained from MS/MS tandem mass spectrometry data. Based on the peptide sequence tag approach.
- MS/MS Ion Search
- Identify fragment ions from uninterpreted MS/MS data of one or more peptides.
The software processes data from mass spectrometers of the following companies:
Important parameters
- Modifications can be specified as fixed or variable.
- Fixed modifications are applied universally to every amino acid residue of the specified type or to the N-terminus or C-terminus of the peptide. The mass for the modification is added to each of the respective residues.
- When variable modifications are specified the program tries to match every different combination of amino acid residues with and without modification. This can increase the number of comparisons dramatically and lead to lower scores and longer search time.
- By setting a taxonomy, a search can be restricted to certain species or groups of species. This will reduce search time and ensure that only relevant protein hits are included.
Scoring
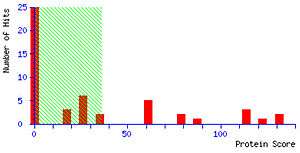
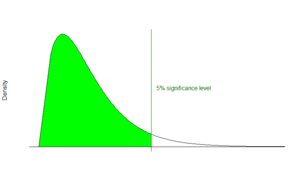
Mascot’s fundamental approach to identifying peptides is to calculate the probability whether an observed match between experimental data and peptide sequences found in a reference database has occurred by chance. The match with the lowest probability of occurring by chance is returned as the most significant match. The significance of the match depends on the size of the database that is being queried. Mascot employs the widely used significance level of 0.05, meaning that in a single test the probability of observing an event at random is less than or equal to 1 in 20. In this light, a score of 10−5 might seem very promising. However, if the database being searched contains 106 sequences several scores of this magnitude would be expected by chance alone because the algorithm carried out 106 individual comparisons. For a database of that size, by applying a Bonferroni correction to account for multiple comparisons, the significance threshold drops to 5*10−8.[1]
In addition to the calculated peptide scores, Mascot also estimates the False Discovery Rate (FDR) by searching against a decoy database. When performing a decoy search, Mascot generates a randomized sequence of the same length for every sequence in the target database. The decoy sequence is generated such that it has the same average amino acid composition as the target database. The FDR is estimated as the ratio of decoy database matches to target database matches. This relates to the standard formula FDR = FP / (FP + TP), where FP are false positives and TP are true positives. The decoy matches are certain to be spurious identifications, but we can't discriminate between true and false positives identified in the target database. FDR estimation was added in response to journals' guidelines on protein identification reports like the ones from Molecular and Cellular Proteomics.[4] Mascot's FDR calculation incorporates ideas from different publications.[5][6]
Alternatives
The most common alternative database search programs are listed in the Mass spectrometry software article. The performance of a variety of mass spectrometry software, including Mascot, can be observed in the 2011 iPRG study. Genome-based peptide fingerprint scanning is another method that compares the peptide fingerprints to the entire genome instead of only annotated genes.
References
- 1 2 Perkins DN, Pappin DJ, Creasy DM, Cottrell JS (December 1999). "Probability-based protein identification by searching sequence databases using mass spectrometry data". Electrophoresis. 20 (18): 3551–67. doi:10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. PMID 10612281.
- ↑ Koenig T, Menze BH, Kirchner M, et al. (September 2008). "Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics". J. Proteome Res. 7 (9): 3708–17. doi:10.1021/pr700859x. PMID 18707158.
- ↑ Pappin DJ, Hojrup P, Bleasby AJ (June 1993). "Rapid identification of proteins by peptide-mass fingerprinting". Curr. Biol. 3 (6): 327–32. doi:10.1016/0960-9822(93)90195-T. PMID 15335725.
- ↑ Bradshaw, R. A. (31 January 2006). "Reporting Protein Identification Data: The next Generation of Guidelines". Molecular & Cellular Proteomics. 5 (5): 787–788. doi:10.1074/mcp.E600005-MCP200.
- ↑ Elias, Joshua E; Haas, Wilhelm; Faherty, Brendan K; Gygi, Steven P (1 September 2005). "Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations". Nature Methods. 2 (9): 667–675. doi:10.1038/nmeth785.
- ↑ Wang, Guanghui; Wu, Wells W.; Zhang, Zheng; Masilamani, Shyama; Shen, Rong-Fong (1 January 2009). "Decoy Methods for Assessing False Positives and False Discovery Rates in Shotgun Proteomics". Analytical Chemistry. 81 (1): 146–159. doi:10.1021/ac801664q.